Flashmob im Zellkern
Bio-News vom 22.06.2021
Der Zellkern ist weit mehr als eine Art Aufbewahrungs-Behälter für Chromosomen: In ihm sitzt auch die komplexe Maschinerie, die Abschriften der gerade benötigten Gene herstellt und in die Zelle entlässt. Manche der daran beteiligten Proteine sind nicht gleichmäßig im Kern verteilt, sondern sammeln sich an bestimmten Stellen.
Fast alle Zellen des Körpers enthalten einen Kern: ein mehr oder weniger kugelförmiges Gebilde, das durch eine Membran vom Rest der Zelle abgegrenzt ist. Jeder Zellkern enthält sämtliche genetischen Informationen des Menschen. Er dient also als eine Art Bibliothek - allerdings eine mit strikten Auflagen: Wenn die Zelle die Bauanleitung für ein Protein benötigt, kann sie nicht einfach die Original-Information entleihen. Stattdessen wird im Kern eine Abschrift davon angefertigt.
Publikation:
Maximilian Schilling, Archana B. Prusty, Björn Boysen, Felix S. Oppermann, Yannick L. Riedel, Alma Husedzinovic, Homa Rasouli, Angelika König, Pradhipa Ramanathan, Jürgen Reymann, Holger Erfle, Henrik Daub, Utz Fischer und Oliver J. Gruss
TOR signaling regulates liquid phase separation of the SMN complex governing snRNP biogenesis
Cell Reports
DOI: 10.1016/j.celrep.2021.109277
Die dazu nötige Maschinerie ist sehr komplex - auch deshalb, weil es sich bei den Abschriften nicht um simple Kopien handelt. Denn Gene enthalten neben wichtigen Informationen auch zahlreiche Passagen mit bedeutungslosem „Müll“. Sie werden beim Herstellen der Abschrift entfernt. Biologen nennen diese redaktionelle Überarbeitung „Splicing“.
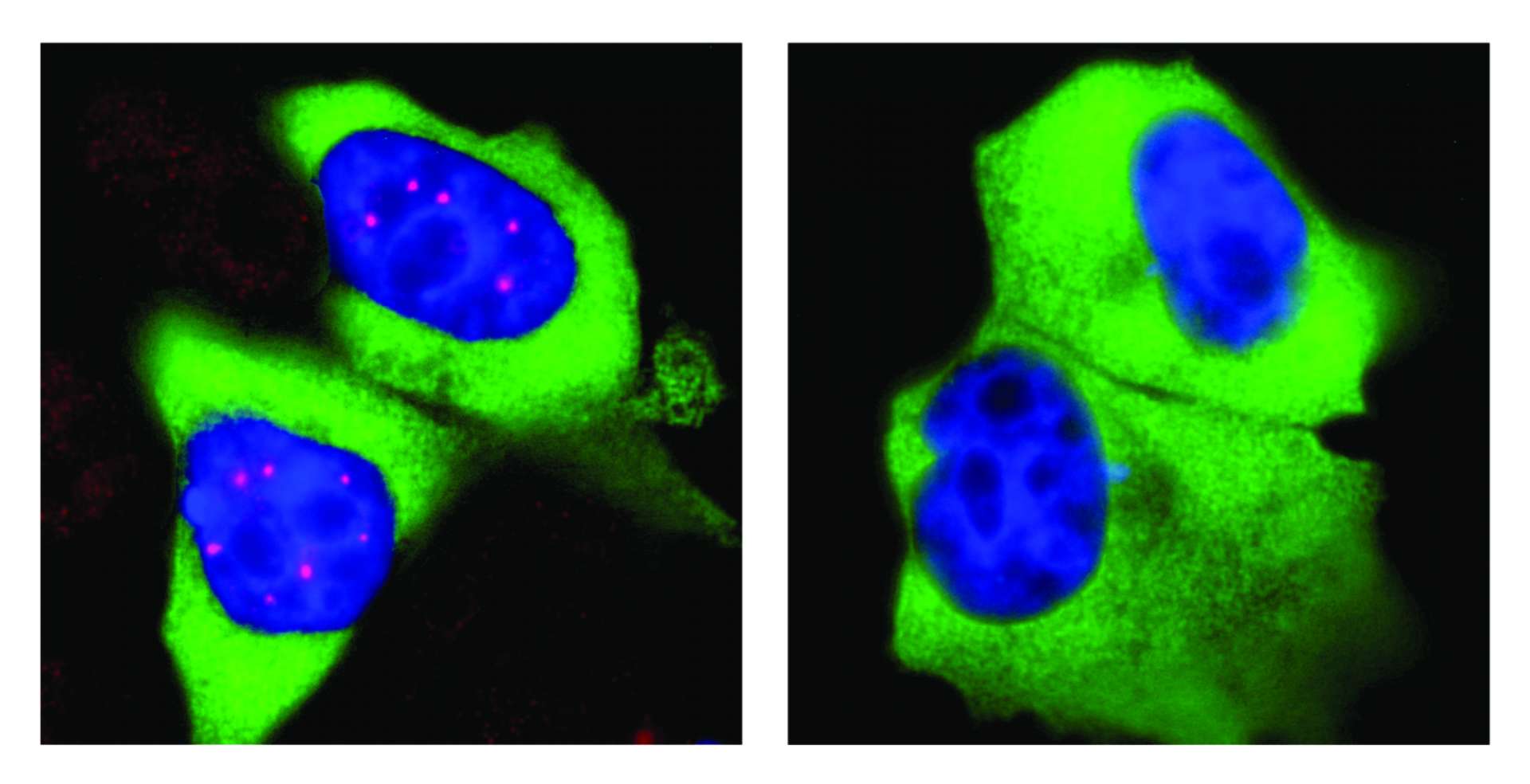
„Eine wichtige Rolle beim Spleißen spielt der SMN-Komplex, eine ‚molekularen Maschine‘ aus mindestens neun verschiedenen Proteinen“, erklärt Prof. Dr. Oliver Gruß vom Institut für Genetik der Universität Bonn, der auch Mitglied im Transdisziplinären Forschungsbereich „Leben und Gesundheit“ der Universität ist. „Interessanterweise sind diese Maschinen nicht gleichmäßig im Zellkern verteilt. Stattdessen sammeln sie sich an bestimmten Stellen, den sogenannten Cajal-Körpern.“ Allerdings gibt es im Zellkern keine Transportmechanismen, die die SMN-Komplexe zu den Cajal-Körpern bringen. Stattdessen haben die SMN-Proteine selbst bestimmte Eigenschaften, die für ihre Zusammenrottung verantwortlich sind. Welche das sind, war bislang unklar.

SMN-Komplexe tragen ungewöhnlich viele Phosphatgruppen
SMN-Komplexe haben eine Auffälligkeit: Sie tragen ungewöhnlich viele Phosphatgruppen - das sind kleine Molekülreste, in deren Zentrum ein Phosphor-Atom sitzt. „Wir haben vermutet, dass diese Phosphorylierung ihre massenhafte Zusammenballung zu Cajal-Körpern fördert“, erläutert Dr. Maximilian Schilling aus der Arbeitsgruppe von Oliver Gruß.
Phosphatgruppen gehören nicht zum eigentlichen Bauplan eines Proteins - sie werden später hinzugefügt und können auch wieder entfernt werden. Oft reguliert die Zelle auf diese Weise die Aktivität des jeweiligen Proteins. Das Anheften der Phosphatgruppe übernehmen dabei bestimmte Enzyme, die Kinasen. „Wir haben nun jede der vielen hundert menschlichen Kinasen einzeln gehemmt und geschaut, wie sich das auf die Bildung der Cajal-Körper auswirkt“, sagt Schilling.
Sie stießen so auf ein Netzwerk von Kinasen, bei deren Hemmung die Cajal-Körper weitgehend verschwanden. Bei weiteren Analysen stellten sie fest, dass ohne diese Kinasen die Phosphorylierung der SMN-Komplexe an bestimmten Stellen stark abnimmt. In der Folge unterbleiben dann die Flashmobs im Zellkern - die Cajal-Körper lösen sich auf. Besonders interessant ist der Befund, weil die identifizierten Kinasen nicht nur das Spleißen regulieren, sondern auch die Übersetzung der so redigierten Genabschriften in Proteine. Es handelt sich also um Enzyme, die für verschiedene Schritte dieses lebenswichtigen Prozesses zentral sind.
Mutation führt zu schwerer Erkrankung
Der SMN-Komplex ist Humangenetikern nicht nur für seine Rolle beim Spleißen bekannt: Einzelne Mutationen in seiner Bauanleitung führen bei Betroffenen zu einer schweren Erkrankung, der spinalen Muskelatrophie. Eines von ca. 6.000 Neugeborenen kommt mit diesem Gendefekt auf die Welt. Die Therapie ist extrem teuer; die Kosten gehen pro Patient in die Millionen. „Ein Teil der Gendefekte, die zur spinalen Muskelatrophie führen, liegen in der Nähe der Phosphorylierungs-Stellen des SMN-Komplexes“, erklärt Gruß. „Bei Betroffenen könnte daher die Anheftung der Phosphatgruppen an diesen Stellen gestört sein, und damit auch die Bildung der Cajal-Körper. Wir vermuten, dass das Spleißen dadurch beeinträchtigt ist, was in der Folge die Erkrankungssymptome hervorruft.“
Womöglich eignen sich die gefundenen Kinasen daher auch als Ansatzpunkt für neue Therapien. Erste Ergebnisse aus Maus-Modellzellen für menschliche spinale Muskelatrophie zeigen, dass sich durch Wirkstoffe, die die Aktivität der Kinasen erhöhen, auch die Bildung der Cajal-Körper verbessert. „Ob diese Wirkstoffe auch krankhafte Veränderungen in einem komplexen Organismus verbessern, ist völlig unklar“, warnt Gruß vor überhöhten Erwartungen. „Dass daraus irgendwann neue Behandlungsmöglichkeiten entstehen werden, ist daher momentan noch Spekulation.“
Beteiligte Institutionen und Förderung
An der Studie waren die Universitäten Bonn, Würzburg und Heidelberg beteiligt. Sie wurde durch die Deutsche Forschungsgemeinschaft (DFG) sowie die US-amerikanische CURE SMA-Foundation gefördert.
Diese Newsmeldung wurde mit Material der Rheinischen Friedrich-Wilhelms-Universität Bonn via Informationsdienst Wissenschaft erstellt.

{kind=link}
{kind=link}