Wie die Evolution arbeitet
Bio-News vom 05.01.2023
Welche genetischen Veränderungen sind für die Entwicklung phänotypischer Merkmale verantwortlich? Diese Frage ist nicht immer leicht zu beantworten. Eine neu entwickelte Methode macht die Suche jetzt deutlich einfacher.
Mit seinen mächtigen Grabschaufeln kann sich der europäische Maulwurf problemlos durch das Erdreich wühlen. Gleiches gilt für den in Australien lebenden Beutelmull. Obwohl die beiden Tierarten weit voneinander entfernt leben, haben sie doch im Laufe der Evolution ähnliche Organe entwickelt – in ihrem Fall für das Graben im Erdboden ideal angepasste Extremitäten.
Publikation:
Fukushima, K., Pollock, D.D.
Detecting macroevolutionary genotype–phenotype associations using error-corrected rates of protein convergence
Nat Ecol Evol 7, 155–170 (2023)
DOI: 10.1038/s41559-022-01932-7
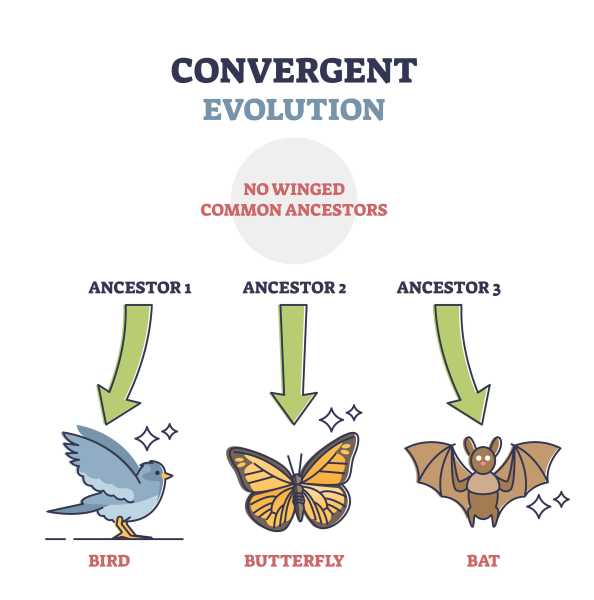
Von „konvergenter Evolution“ spricht die Wissenschaft in solchen Fällen, wenn Tier-, aber auch Pflanzenarten unabhängig voneinander Merkmale entwickeln, die die gleiche Gestalt und Funktion haben. Beispiele gibt es dafür viele: So besitzen Fische Flossen, genauso wie Wale, die allerdings zu den Säugetieren zählen. Vögel und Fledermäuse verfügen über Flügel, und wenn es darum geht, sich mit Hilfe giftiger Substanzen gegen Angreifer zu wehren, haben viele Lebewesen, von Quallen über Skorpione bis zu Insekten, alle das gleiche Instrument entwickelt: den Giftstachel.


Identische Merkmale trotz fehlender Verwandtschaft
Klar, dass sich Wissenschaftlerinnen und Wissenschaftler weltweit dafür interessieren, welche Veränderungen im Erbgut der jeweiligen Arten dafür verantwortlich sind, dass sich bei ihnen identische Merkmale entwickeln konnten, obwohl unter ihnen keine verwandtschaftlichen Beziehungen bestehen.
Die Suche danach gestaltet sich schwierig: „Solche Merkmale – wir sprechen von Phänotypen – sind natürlich immer in Genomsequenzen kodiert“, sagt der Pflanzenphysiologe Dr. Kenji Fukushima von der Julius-Maximilians-Universität (JMU) Würzburg. Mutationen – also Veränderungen im Erbgut – können die Auslöser für die Entwicklung neuer Merkmale sein.
Allerdings führen genetische Veränderungen selten zu einer phänotypischen Evolution, da die zugrunde liegenden Mutationen weitgehend zufällig und neutral sind. Somit sammeln sich in der extremen Zeitskala, in der sich evolutionäre Prozesse vollziehen, eine gewaltige Menge an Mutationen an, was die Entdeckung phänotypisch wichtiger Veränderungen äußerst schwierig macht.
Neuartige Metrik der molekularen Evolution
Jetzt ist es Fukushima gemeinsam mit seinem Kollegen David D. Pollock von der University of Colorado (USA) gelungen, eine Methode zu entwickeln, die bei der Suche nach den genetischen Grundlagen phänotypischer Merkmale deutlich bessere Ergebnisse erzielt als die bislang verwendeten Methoden. In der aktuellen Ausgabe der Fachzeitschrift Nature Ecology & Evolution stellen sie ihren Ansatz vor.
„Wir haben eine neuartige Metrik der molekularen Evolution entwickelt, mit der sich die Rate der konvergenten Evolution in proteinkodierenden DNA-Sequenzen genau darstellen lässt“, beschreibt Fukushima das wesentliche Ergebnis der jetzt veröffentlichten Arbeit. Diese neue Methode könne auf einer evolutionären Zeitskala von Hunderten von Millionen Jahren aufzeigen, welche genetischen Veränderungen mit den Phänotypen von Organismen verbunden sind. Damit biete sie die Möglichkeit, das Verständnis dafür zu erweitern, wie Veränderungen in der DNA zu phänotypischen Innovationen führen, die eine große Artenvielfalt hervorbringen.
Gewaltiger Datenschatz als Grundlage
Eine zentrale Entwicklung in den Lebenswissenschaften bildet die Grundlage von Fukushimas und Pollocks Arbeit: die Tatsache, dass in den vergangenen Jahren immer mehr Genomsequenzen vieler Lebewesen quer durch die Artenvielfalt entschlüsselt und damit einer Analyse zugänglich gemacht wurden. „Damit wurde es möglich, auf einer makroevolutionären Ebene die Zusammenhänge von Geno- und Phänotypen in großem Maßstab zu untersuchen“, sagt Fukushima.
Da jedoch viele molekulare Veränderungen nahezu neutral seien und sich nicht auf irgendwelche Merkmale auswirken, bestehe bei der Interpretation der Daten häufig die Gefahr einer „falsch-positiven Konvergenz“ – soll heißen: Das Ergebnis sagt einen Zusammenhang zwischen einer Mutation und einem bestimmten Merkmal voraus, der in Wirklichkeit jedoch nicht existiert. Darüber hinaus könnten auch methodische Verzerrungen für solche falsch-positiven Konvergenzen verantwortlich sein.
Zusammenhänge über Millionen von Jahren
„Um dieses Problem zu überwinden, haben wir den Rahmen erweitert und eine neue Metrik entwickelt, die die fehlerbereinigte Konvergenzrate der Proteinevolution misst“, erklärt Fukushima. Damit sei es möglich, die natürliche Selektion von genetischem Rauschen und phylogenetischen Fehlern in Simulationen und realen Beispielen zu unterscheiden. Erweitert um einen heuristischen Algorithmus ermögliche dieser Ansatz die bidirektionale Suche nach Genotyp-Phänotyp-Assoziationen, selbst in Linien, die sich über Hunderte von Millionen Jahren auseinanderentwickelt haben.
Wie gut die von ihnen entwickelte Metrik funktioniert, haben die beiden Wissenschaftler anhand von über 20 Millionen Zweigkombinationen bei Wirbeltiergenen untersucht. In einem nächsten Schritt wollen sie diese Methode auf fleischfressende Pflanzen anwenden. Ziel ist es, die genetischen Grundlagen zu entziffern, die dafür mitverantwortlich sind, dass diese Pflanzen Beute anlocken, fangen und verdauen können.
Diese Newsmeldung wurde mit Material der Julius-Maximilians-Universität Würzburg via Informationsdienst Wissenschaft erstellt.

{kind=link}
{kind=link}
{kind=link}
_(cropped).jpg){kind=link}