Hämoglobin
- Wikipedia:Vorlagenfehler/Vorlage:Cite journal/temporär
- Wikipedia:Vorlagenfehler/Vorlage:Cite book/temporär
- Seiten mit Skriptfehlern
- Zellbestandteil
- Sauerstofftransporter
- Blutbild
- Pigment (Biologie)
- Porphyrinkomplex
- Proteinkomplex
- Erbkrankheit-assoziiertes Protein
- Wikipedia:Artikel-Feedback/Zusätzliche Artikel
| Hämoglobin α-Untereinheit | ||
|---|---|---|
|
| ||
| Hb-Dimer: vorne α-Untereinheit als Cartoon-Modell mit Häm (rot, als VDW-Modell), hinten β-Untereinheit als Stäbchen, nach PDB 1GZX | ||
| Vorhandene Strukturdaten: UniProt-Eintrag | ||
| Masse/Länge Primärstruktur | 16 kDa, 141 Aminosäuren (je Untereinheit) | |
| Kofaktor | Häm | |
| Bezeichner | ||
| Gen-Name(n) | HBA1, HBA2 | |
| Externe IDs | OMIM: 141800 UniProt: P69905 | |
| Vorkommen | ||
| Homologie-Familie | Beta-2 Globin | |
| Übergeordnetes Taxon | Wirbeltiere | |

| Hämoglobin β-Untereinheit | ||
|---|---|---|
|
| ||
| Kugelmodell der Häm-Tasche der Hämoglobin β-Untereinheit mit Häm, Eisen (grün) und Disauerstoff, nach PDB 1GZX | ||
| Vorhandene Strukturdaten: UniProt-Eintrag | ||
| Eigenschaften des menschlichen Proteins | ||
| Masse/Länge Primärstruktur | 146 Aminosäuren | |
| Kofaktor | Häm | |
| Bezeichner | ||
| Gen-Name | HBB | |
| Externe IDs | OMIM: 141900 UniProt: P68871 MGI: 96021 | |
| Vorkommen | ||
| Homologie-Familie | Beta-2 Globin | |
| Übergeordnetes Taxon | Wirbeltiere | |

Hämoglobine (von griech. αἷμα haíma ‚Blut‘ und lat. globus ‚Kugel‘) (Hb) sind eisenhaltige, sauerstofftransportierende Proteine, die in den roten Blutkörperchen und ihren Varianten von Wirbeltieren, einiger Anneliden, Mollusken sowie weniger Crustaceen und Insekten gefunden werden. Sie nehmen den Sauerstoff in der Lunge bzw. den Kiemen auf und verteilen ihn im Körper. Die Bindung des Sauerstoffs erfolgt an einem Eisenkomplex des Protoporphyrins IX (Häm). Wirbeltier-Hämoglobine sind Proteinkomplexe aus vier Untereinheiten (2 α, 2 β), die jeweils eine Häm-Gruppe, die in ein Globin eingebettet ist, enthalten. Die Häm-Gruppe ist außerdem für die rote Farbe des Hämoglobins verantwortlich.
Geschichte
Das Sauerstofftransportprotein Hämoglobin wurde 1840 von Hünefeld entdeckt.[1] 1851 beschrieb Otto Funke die Kristallisation von Hämoglobin durch Verdünnen von Tierblut mit Wasser, Ethanol oder Diethylether und anschließendem langsamen Verdampfen des Lösungsmittels aus der erhaltenen Proteinlösung („Funkesche Kristalle“).[2] Über die reversible Oxygenierung des Hämoglobins wurde 1866 erstmals von Felix Hoppe-Seyler berichtet.[3] Von ihm stammt auch der Name Hämoglobin. Das Hämoglobin ist eines der bestuntersuchten Proteine, seine Struktur wurde als eine der ersten überhaupt von Max Perutz et al 1959 mit Hilfe der Röntgenkristallographie ermittelt.[4][5][6][7] Für diese Arbeiten bekam er 1962 zusammen mit John Kendrew den Nobelpreis für Chemie.
Struktur
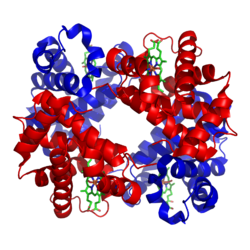
Bändermodell (Untereinheit α und β, rot bzw. blau) von Hämoglobin A mit der prosthetischen Gruppe Häm (Stäbchenmodell, grün) nach PDB 1GZX

Strukturformel des Häm b


Säuger-Hämoglobine bestehen aus vier Untereinheiten, je zwei vom α- und zwei vom β-Typ. In jede dieser Untereinheiten ist jeweils eine prosthetische Gruppe, an der die Sauerstoffbindung stattfindet, eingebettet. Ein Hämoglobinkomplex-Molekül kann also vier Sauerstoffmoleküle binden. Die prosthetische Gruppe der sauerstofffreien Form ist ein Eisen(II)-Komplex des Protoporphyrins IX, welches die vier äquatorialen Positionen des Eisenions besetzt. Das Eisenion befindet sich in einem high-spin-Zustand und ist daher etwas zu groß, um in das Loch des Porphyrins zu passen. Es befindet sich also etwas unterhalb der Ringebene. Dieses Häm b ist über die axiale Position des Eisenions auf der Unterseite über einen proximalem Histidinrest an die Proteinmatrix gebunden. Die zweite axiale Position auf der Oberseite ist unbesetzt und steht für die Anbindung des Sauerstoffmoleküls zur Verfügung.
Posttranslationale Modifikationen
Neben verschiedenen seltenen Modifikationen einzelner Aminosäuren in den Hämoglobin-Untereinheiten des Menschen tritt häufig die Glykation beider Untereinheiten an speziellen Aminosäuren auf. Dies ist die Folge einer hohen Glucosekonzentration im Blut und kann daher in der Labordiagnostik verwendet werden, um den durchschnittlichen Blutzuckerspiegel der letzten Monate zu ermitteln.
Bei der Glykation des Hämoglobins wird Glucose kovalent an Lysin-8, -17, -41, -62 der α-Untereinheit, sowie Valin-2, Lysin-9, -18, -67, -121 oder -145 der β-Untereinheit gebunden. Ist ein glykiertes β-Hämoglobin an Valin-2 modifiziert, und hat sich der Glucoserest über ein Aldimin und eine Amadori-Umlagerung zu einem stabilen Ketoamin gewandelt, wird es als HbA1c bezeichnet.[8][9]
Methämoglobin
Methämoglobin ist eine deaktivierte, nicht sauerstoffaffine Form des Hämoglobins, in dem das Eisenion sich in der Oxidationsstufe III statt II befindet. Das NADH-abhängige Enzym Methämoglobinreduktase (Diaphorase I) ist dafür verantwortlich, Methämoglobin wieder in Hämoglobin zu überführen. In der Regel liegen beim Menschen zwei Prozent des Hämoglobins als Methämoglobin vor. Ein höherer Anteil ist entweder genetisch bedingt oder eine Folge einer Vergiftung. In Abhängigkeit vom Methämoglobinlevel kann dies zu Gesundheitsproblemen führen.
Sauerstofftransport
Leistungsfähigkeit

Hämoglobin ist ein globuläres Protein mit sehr guter Löslichkeit in Wasser (Löslichkeit bis zu 5 mmol/l Hämoglobin (34 %)). 1 g Hb kann in vitro 1,389 ml Sauerstoff binden, in vivo jedoch nur 1,34 ml (Hüfnersche Zahl), somit können 100 ml Blut, die etwa 15 g Hb enthalten, bei 100-prozentiger Sättigung bis zu 15 × 1,34 ml = 20,1 ml Sauerstoff aufnehmen.
Auffällig ist der sigmoidale (S-förmige) Verlauf der Bindungskurve. Normalerweise würde man erwarten, dass die Sauerstoffbeladung mit steigendem Sauerstoffpartialdruck wie beim Myoglobin zunächst stark und dann immer langsamer zunimmt (hyperbolischer Verlauf). Für Hämoglobin verläuft die Sauerstoffbindungskurve im Bereich des in der Lunge herrschenden Sauerstoffpartialdrucks ungewöhnlich flach und im Bereich des im Gewebe herrschenden Sauerstoffpartialdrucks ungewöhnlich steil. Der flache Verlauf der Bindungskurve im Endteil verhindert einen stärkeren Abfall der Sauerstoffsättigung im Alter, bei Lungenfunktionsstörungen und in Höhenlagen, und der steilere Verlauf im Mittelteil sorgt dafür, dass bei einem sinkenden venösen Sauerstoffpartialdruck viel Sauerstoff abgegeben wird.[11][12]
Normbereich
Für das beim Erwachsenen überwiegende sogenannte „adulte Hämoglobin“ (siehe unten) wurde ein Normbereich festgelegt. Für Kinder gelten andere Normwerte.
Als Normalbereich wird der Bereich bezeichnet, in dem die Hb-Werte von 96 Prozent aller gesunden Menschen liegen.
| g/dl (alte Einheit) |
mmol/l* (SI-Einheit) | |
|---|---|---|
| Männer | 13,5–17,5 | 8,7–11,2 |
| Frauen | 12–16 | 7,5–9,9 |
| Neugeborene | 19 |
Ein erhöhter Hämoglobin-Wert bedeutet meistens auch eine erhöhte Erythrozyten-Anzahl (Polyglobulie) und kann z. B. bei Aufenthalt in großen Höhen (Sauerstoffmangel) oder durch Flüssigkeitsverlust auftreten. Bei unklarer Ursache ist auch abzuklären, ob es sich bei stark erhöhten Werten um die Erkrankung Polycythaemia vera handelt.
Ein verringerter Hämoglobin-Wert wird als Anämie bezeichnet, eine verminderte Beladung der roten Blutkörperchen mit Hämoglobin als Hypochromasie.
Ein erhöhter/verringerter Hämoglobin-Wert ist immer abhängig vom Normalwert. Liegt der Normalwert bei 11,2 mmol/l, dann kann ein Wert von 8,7 mmol/l schon zu Symptomen von Anämie führen. Liegt der Normalwert bei 9 mmol/l, dann treten bei 8,7 mmol/l noch keine Symptome auf.
Die Höhe des Hämoglobinwertes ist maßgeblich bei der Zulassung zur Blutspende. Männer müssen einen Mindestwert von 13,5 g/dl, Frauen einen von 12,5 g/dl aufweisen, um vom Spendearzt zugelassen zu werden. Bestimmt wird der HB-Wert mittels elektronisch messender HB-Photometer. Aktuelle Informationen des DRK Blutspendedienstes sagen aus, dass Männer mit einem HB von >18,0 g/dl nicht mehr zur Spende zugelassen werden (12/2006). Dies ist jedoch nicht in allen Bundesländern der Fall, bei erhöhten HB-Wert wird weitere Flüssigkeitsaufnahme vor der Spende empfohlen.
Die Bestimmung des Hämoglobingehalts basiert auf dem Nachweis der Hämgruppen des Proteins. Daher wird als molare Hämoglobinkonzentration traditionell (so auch hier) die Konzentration der einzelnen Untereinheiten angegeben. Da ein Hämoglobinkomplex-Molekül aus 4 Untereinheiten mit je einer Hämgruppe besteht, müssen die in mmol/l angegebenen Werte durch 4 geteilt werden, um die wahre Konzentration des Hämoglobins zu erhalten. Die in g/dl angegebenen Werte bleiben unverändert.
Sauerstoffbindung durch Hämoglobine auf molekularer Ebene

Bei der Bindung des Disauerstoffmoleküls wird das Eisenzentrum zur Oxidationsstufe III oxidiert. Das Sauerstoffmolekül wird zum Superoxid reduziert und bindet an das Eisenzentrum (Formulierung nach Weiss). Pauling formuliert dagegen einen Fe(II)-Disauerstoffkomplex. In der Nähe des Eisenzentrums befindet sich ein distales Histidin, welches das gebundene Sauerstoffmolekül über eine Wasserstoffbrücke stabilisiert. Das Eisenzentrum geht durch die Sauerstoffbindung in einen low-spin-Zustand über. Dabei verringert sich seine Größe und es rutscht in den Porphyrinring hinein.
Kooperativer Effekt bei der Sauerstoffbindung
Ein Hämoglobinmolekül kann vier Sauerstoffmoleküle binden. Aus rein statischen Erwägungen wäre zu erwarten, dass das Bestreben, weitere Sauerstoffmoleküle zu binden, mit jedem bereits gebundenen Sauerstoffmolekül abnimmt. Untersuchungen haben jedoch gezeigt, dass das Gegenteil der Fall ist und die Sauerstoffaffinität mit steigender Beladung zunimmt (positive Kooperativität).[13][14]
pH-Wert-Abhängigkeit der Sauerstoffbindung
Das Gleichgewicht zwischen R- und T-Form ist pH-abhängig (Bohr-Effekt) und wird bei einem niedrigen pH-Wert durch Protonierung zugunsten der weniger sauerstoffaffinen T-Form verschoben. Denselben Effekt hat ein hoher CO2 -Partialdruck durch eine reversible Carboxylierung der Untereinheiten. Dies führt dazu, dass hohe Protonen- und Kohlenstoffdioxid-Konzentrationen, wie sie z.B. durch Zellatmung und Milchsäuregärung im arbeitenden Muskel herrschen, eine vollständige Entladung des Hämoglobins begünstigen.
Konkurrenz zwischen Sauerstoff und Kohlenstoffmonoxid
Kohlenstoffmonoxid (CO) ist sehr giftig, da es mit dem Sauerstoff um die axialen Koordinationsstellen der Eisenzentren konkurriert. An das freie Häm b bindet Kohlenstoffmonoxid 25000 Mal stärker als Disauerstoff, im Hämoglobin dagegen nur etwa 200 Mal. Die Ursache für die verringerte CO-Affinität ist, dass aufgrund des Raumbedarfes des distalen Histidins, die vom Kohlenstoffmonoxid bevorzugte lineare Fe-CO-Koordination nicht möglich ist. Einmal gebunden, kann CO z. B. lediglich durch Sauerstoffdruckbehandlung in einer Druckkammer verdrängt werden – es blockiert also Bindungsstellen für Sauerstoff. Bei starken Rauchern sind bis zu 10 Prozent des Hämoglobins mit CO besetzt.
Effekte, welche die Sauerstoffbindung beeinflussen
Die Sauerstoffbindungskurve wird auf der Abszisse nach rechts verschoben durch:
- Temperaturanstieg
- Absinken des pH-Werts
- Steigerung der Konzentration von 2,3-Bisphosphoglycerat in den Erythrozyten
- Steigerung der Konzentration von Kohlenstoffdioxid
Die Rechtsverschiebung führt dazu, dass das Hämoglobin leichter Sauerstoff abgibt. Ein Beispiel: Ein arbeitender Muskel verbraucht sehr viel Sauerstoff für die Kontraktion. Da er die Energie zum Teil in Wärme umsetzt, steigt in der arbeitenden Muskulatur die Temperatur an. Außerdem setzt er Milchsäure frei, der pH-Wert sinkt. Durch den gesteigerten Stoffwechsel entsteht vermehrt Kohlenstoffdioxid: durch die lokalen Effekte kann die Muskulatur mehr Sauerstoff aus dem Blut entnehmen.
- Die Muskulatur verfügt über Myoglobin (siehe unten), das eine höhere Affinität zu Sauerstoff besitzt (Sauerstoff stärker anzieht). Es dient als Sauerstoff-Speicher.
Die Sauerstoffbindungskurve wird auf der Abszisse nach links verschoben durch:
- Temperaturabnahme
- Anstieg des pH-Werts
- Absinken der Konzentration von 2,3-Bisphosphoglycerat in den Erythrozyten
- Absinken der Konzentration von Kohlenstoffdioxid
Die Linksverschiebung führt dazu, dass Hämoglobin Sauerstoff stärker bindet. Dies macht man sich z. B. bei Herzoperationen zu nutze, indem man den Patienten unterkühlt, um sein Blut maximal mit Sauerstoff zu sättigen. Tiere, die Winterschlaf halten, profitieren ebenfalls von diesem Effekt. In den Lungen wird ein Teil des Kohlendioxids im Zuge der Ausatmung abgegeben – das Hb kann wieder leichter mit Sauerstoff beladen werden.
Das im Rapoport-Luebering-Zyklus (Nebenweg der Glycolyse) durch das Enzym Bisphosphoglyceratmutase gebildete 2,3-Bisphosphoglycerat ist das wichtigste Intermediat der Glykolyse in den roten Blutkörperchen. Es bindet an Hämoglobin und verursacht einen allosterischen Effekt; die Abnahme der Affinität des Hämoglobins zum Sauerstoff. Es ist lebenswichtig, um die Abgabe von Sauerstoff im Organismus zu ermöglichen. Unter physiologischen Bedingungen liegt 2,3-BPG in den roten Blutkörperchen etwa in derselben Konzentration wie Hämoglobin vor. Eine Erhöhung der 2,3-BPG-Konzentration ist z.B. bei der Höhenanpassung zu beobachten. Sinn dieser Regulation ist folgender:
Ist die Sauerstoffsättigung im Blut durch die „dünne Luft“ in großer Höhe vermindert, gibt Hb den gebundenen Sauerstoff an die Verbraucher schlechter ab als bei hoher Sättigung (siehe Bindungskurve). Trotzdem muss die Versorgung aller Organe mit O2 gewährleistet sein. Hb muss also weniger affin zum Sauerstoff werden, um die Peripherie ausreichend zu versorgen.
Hämoglobin-Typen
Es gibt beim Menschen vier leicht verschiedene Hämoglobine, wobei mit Hämoglobin im Allgemeinen der Haupttyp A1 (HbA1 oder auch einfach HbA) bezeichnet wird. Außerdem gibt es noch Hämoglobin A2 (HbA2), sowie die fetalen Hämoglobine HbF und Gower-2. Diese vier Proteinkomplexe unterscheiden sich in den Protein-Untereinheiten, aus denen sie aufgebaut sind: HbA1 besteht aus zwei α- und zwei β-, HbF aus zwei α- und zwei γ-, HbA2 aus zwei α- und zwei δ-, sowie Gower-2 aus zwei α- und zwei ε-Untereinheiten.

Durch Genduplikationen des Globin-Gens sind unterschiedliche Untereinheiten des Hämoglobins entstanden, welche unterschiedliche Affinitäten für Sauerstoff besitzen. Die Kombinationen dieser Untereinheiten als Tetramer werden je nach Sauerstoffbedarf zu unterschiedlichen Zeitpunkten synthetisiert, beispielsweise um als Fötus im Mutterleib Sauerstoff aus dem mütterlichen Blut zu erhalten.
Embryonale Hämoglobine
Während der Schwangerschaft muss Sauerstoff durch die Plazenta (Mutterkuchen) zum Fötus transportiert werden. Das ungeborene Kind besitzt einen anderen Hämoglobin-Typ, das HbF, das Sauerstoff mit wesentlich höherer Affinität bindet. Außerdem ist der Hämatokrit im Vergleich zur Mutter stark erhöht. Auf diese Weise gelangt durch die Nabelschnur genug Sauerstoff zum Fötus.
Die embryonalen Hämoglobine werden in der Embryonalphase, d.h. während der ersten 8 Wochen nach Befruchtung im Dottersack gebildet und tragen Eigennamen:
- Gower 1 (ζ2ε2) ("zeta-epsilon")
- Gower 2 (α2ε2) ("alpha-epsilon")
- Portland (ζ2γ2) ("zeta-gamma")
Fetale Hämoglobine
Als fetales Hämoglobin wird das während der Fetalperiode (ab der 9. Woche nach Befruchtung bis zur Geburt) vorwiegend gebildete Hämoglobin F (HbF) bezeichnet. Die Synthese des fetalen Hämoglobins beginnt schon in der vorangehenden Embryonalphase und wird auch nicht sofort nach Geburt gestoppt, sondern hält noch einige Monate an. Bildungsort sind Leber und Milz. Es hat eine viel höhere Sauerstoffaffinität als adultes Hämoglobin, um den Sauerstoff aus dem mütterlichen Blut aufzunehmen.
- Hämoglobin F (α2γ2) - Im Fötus das dominierende Hämoglobin, bei gesunden Erwachsenen nur in Spuren nachweisbar
Adulte Hämoglobine
Als adulte Hämoglobine gelten Hämoglobin A1 (HbA1) und Hämoglobin A2 (HbA2). Die Synthese der adulten Hämoglobine beginnt schon im Fetus und ersetzt dann in den ersten Monaten nach Geburt das fetale Hämoglobin. Bildungsort ist das Knochenmark. Beim gesunden Erwachsenen ist fetales Hämoglobin nur in Spuren nachweisbar.
- Hämoglobin A1 (α2β2) - 98 %
- Hämoglobin A2 (α2δ2) - 2 %.
Abbau von Hämoglobin
Wenn die roten Blutkörperchen das Ende ihres Lebens (etwa 120 Tage) erreicht haben, werden sie in den mononukleären Phagozyten hauptsächlich in der Milz (und bei hohem Anfall von abzubauendem Hämoglobin auch in der Leber und im Knochenmark) abgebaut. Der Abbauprozess beginnt in der Milz und wird in der Leber fortgesetzt. Zuerst wird der Globinanteil vom Häm getrennt und zu Aminosäuren degradiert. Das Häm wird über eine Cytochrom-P450-abhängige Oxygenase (Hämooxygenase) zu Biliverdin gespalten, wobei Eisen (Fe2+) und Kohlenmonoxid frei werden. Das Eisen wird von den Makrophagen an das im Blut vorhandene Transportprotein Transferrin abgegeben. Die Biliverdin-Reduktase wandelt schließlich das grünliche Biliverdin in Bilirubin um. Dieses wird ins Blut abgegeben und an Albumin gekoppelt (da es schlecht wasserlöslich ist) und zur Leber transportiert. Hier wird das Bilirubin zweifach mit Glucuronsäure konjugiert und somit löslich gemacht – es kann nun mit der Galle in den Darm abgegeben und über den Stuhl ausgeschieden werden. Im Darm sorgen Bakterien dafür, dass die Glucuronsäure teilweise wieder gespalten wird, und auf diese Weise das orangefarbene Bilirubin reduziert wird zu farblosem Urobilinogen und zu braunem Sterkobilinogen. Ein geringer Anteil dieses reduzierten Bilirubins wird wieder über den Darm aufgenommen und über die Niere ausgeschieden (gelbe Farbe des Urins). Verschiedene Lebererkrankungen wie die Leberentzündung (Hepatitis) oder Abflussbehinderungen in der Gallenblase (Gallensteine) führen zu einer erhöhten Bilirubinkonzentration. Bilirubin ist der Farbstoff, der bei Einlagerung in die Haut zur so genannten Gelbsucht (Ikterus = Gelbfärbung der Haut und Lederhaut (Sclera) der Augen) führt.
Rolle bei Krankheiten
Mutationen im HBA1-Gen können zu Defekten der α-Untereinheit und diese zu Heinz-Körper-Anämie und zur α-Thalassämie führen. Mutationen im HBB-Gen können die Ursache für Heinz-Körper-Anämie, β-Thalassämie und Sichelzellenanämie werden. Manche Varianten der HBG1/HBG2-Gene können Neugeborenengelbsucht verursachen.[15]
Reduzierte Hämoglobinwerte, mit oder ohne Reduktion der Zahl von roten Blutkörperchen, führen zu den Symptomen einer Anämie. Es gibt viele Ursachen für eine Anämie, wobei Eisenmangel der häufigste Grund in der westlichen Welt sein dürfte. Durch Eisenmangel wird die Häm-Synthese gehemmt. Als Folge sind die roten Blutkörperchen hypochromisch (ohne die rote Farbe) und mikrozytisch (kleiner als normal).
Bei einer Hämolyse (verstärkter Abbau von roten Blutkörperchen) tritt eine Gelbsucht auf, verursacht durch das Hämoglobin-Metabolit Bilirubin.
Eine Gruppe von genetischen Defekten, bekannt als Porphyrien, führen zu einer Störung der Hämsynthese. Durch die Anreicherung von Häm-Vorstufen kommt es unter anderem zu Lichtempfindlichkeit, Abdominalschmerzen und neurologischen Problemen.
Mutationen in den Globinketten sind mit verschiedenen Hämoglobinopathieen verbunden, wie die Sichelzellenanämie und Thalassämie.
Die Erreger der Malaria spalten Hämoglobin in von ihnen infizierten roten Blutkörperchen, um daraus Proteine für ihren eigenen Stoffwechsel zu gewinnen. Aus dem Häm entsteht dabei Hämozoin, das vom Parasiten kristallisiert wird und unter dem Mikroskop in den infizierten Erythrozyten als Pigment erkennbar ist. Das Malaria-Medikament Chloroquin hemmt diese Kristallisierung und der Parasit wird durch das Häm vergiftet.
Nachweis
Der Nachweis von Hämoglobin erfolgt durch den Teichmann-Test, bei dem Blut vorsichtig mit Kochsalz und Eisessig erwärmt wird, wobei sich Hämin (Teichmann-Kristalle) abscheidet, oder mit der Luminolreaktion, bei der eine Lösung aus Luminol sowie Natronlauge und eine Lösung aus Wasserstoffperoxid verwendet wird. Diese Reaktion findet nur in Anwesenheit eines Katalysators statt, im Nachweisfalle von Hämoglobin wäre dieser das Eisen-(II)-Ion im Häm-Komplex.
Siehe auch
Andere sauerstoffbindende Proteine
Hämoglobin ist nicht der einzige Sauerstofftransporter in der Natur.
- Myoglobin übernimmt in den Muskeln von Wirbeltieren (inklusive Mensch) den Sauerstoff vom Hämoglobin. Sein aktives Zentrum ist ebenfalls ein Häm b, allerdings ist es im Unterschied zum Hämoglobin nicht tetra- sondern monomer.
- Hämocyanin ist der zweithäufigste Sauerstofftransporter in der Tierwelt und wird im Blutplasma vieler Arthropoden und Mollusken gefunden. Seine prosthetische Gruppe ist ein zweikerniger Kupfer(I)-Komplex.
- Hämerythrin wird von einigen marinen Invertebraten sowie wenigen Arten der Anneliden verwendet, um Sauerstoff in ihren Blutzellen zu transportieren. Sein aktives Zentrum wird von zwei Eisen(II)-Ionen gebildet.
Weiteres
- Hämolyse
Hämoglobin in Kunst und Musik

Der Deutsch-Amerikanische Künstler Julian Voss-Andreae hat 2005 eine Skulptur erschaffen, die auf der Struktur des Hämoglobins beruht[16][17]. Das absichtsvolle Rosten des Werks reflektiert die Oxidation des Eisenatoms im Häm.
Die britische Rockband Placebo nahm ein Lied mit dem Titel Haemoglobin auf. Der französische Rapper MC Solaar veröffentlichte im Jahr 1994 eine erfolgreiche Single mit dem Title La concubine de l'hémoglobine. Die deutsche Melodic-Death-Metalband Deadlock hat auf ihrem Album "The Arrival" ein Lied mit dem Namen "Killing The Time With Haemoglobin" mit einer Länge von 11 Minuten.
Einzelnachweise
- ↑ Hünefeld F.L.: Die Chemismus in der thierischen Organization. Leipzig, 1840.Vorlage:Cite book/Meldung
- ↑ A NASA Recipe For Protein Crystallography (PDF) In: Educational Brief. National Aeronautics and Space Administration. Abgerufen am 15. September 2012.
- ↑ Hoppe-Seyler F: Über die oxydation in lebendem blute. In: Med-chem Untersuch Lab. 1. Jahrgang, 1866, S. 133–140.
- ↑ M.F. Perutz, M.G. Rossmann, A.F. Cullis, H. Muirhead, G. Will, A.C.T. North: Structure of H. In: Nature. 185. Jahrgang, Nr. 4711, 1960, S. 416–422, doi:10.1038/185416a0, PMID 18990801.
- ↑ Perutz MF: Structure of hemoglobin. In: Brookhaven symposia in biology. 13. Jahrgang, November 1960, ISSN 0068-2799, S. 165–83, PMID 13734651.
- ↑ Perutz MF, Miurhead H, Cox JM, et al: Three-dimensional Fourier synthesis of horse oxyhaemoglobin at 2.8 A resolution: (1) x-ray analysis. In: Nature. 219. Jahrgang, Nr. 5149, Juli 1968, S. 29–32, doi:10.1038/219029a0, PMID 5659617.
- ↑ Perutz MF, Muirhead H, Cox JM, Goaman LC: Three-dimensional Fourier synthesis of horse oxyhaemoglobin at 2.8 A resolution: the atomic model. In: Nature. 219. Jahrgang, Nr. 5150, Juli 1968, S. 131–9, PMID 5659637.
- ↑ Canterbury Scientific: HbA1c and Glycated Hemoglobin
- ↑ UniProt P69905, UniProt P68871
- ↑ J. M. Berg, J. L. Tymoczko, L. Stryer: Biochemie. 6. Auflage. Spektrum Akademischer Verlag, Elsevier GmbH, München 2007; S. 208ff; ISBN 978-3-8274-1800-5
- ↑ Robert F. Schmidt, Gerhard Thews (Hrsg.): Physiologie des Menschen. 25. Auflage. Springer-Verlag, Berlin 1993, ISBN 3-541-02636-7, S. 616–620.
- ↑ Erich Schütz, Heinz Caspers, Erwin-Josef Speckmann (Hrsg.): Physiologie. 16. Auflage. Urban & Schwarzberg, München 1982, ISBN 3-540-57104-3, S. 86–87.
- ↑ Adair GS: The hemoglobin system. VI. The oxygen dissociation curve of hemoglobin. In: J Biol Chem 1925;63:529–545.
- ↑ D. E. Koshland, G. Némethy, D. Filmer: Comparison of experimental binding data and theoretical models in proteins containing subunits. In: Biochemistry. Band 5, Nummer 1, Januar 1966, S. 365–385, ISSN 0006-2960. PMID 5938952.
- ↑ UniProt P69905, UniProt P68871, UniProt P69891
- ↑ Constance Holden: Blood and Steel. In: Science. 309. Jahrgang, Nr. 5744, 30. September 2005, S. 2160, doi:10.1126/science.309.5744.2160d (sciencemag.org [PDF]).
- ↑ Moran L, Horton RA, Scrimgeour G, Perry M: Principles of Biochemistry. Pearson, Boston, MA 2011, ISBN 0-321-70733-8, S. 127.
Literatur
- M. F. Perutz: Stereochemistry of cooperative effects in haemoglobin. In: Nature. Band 228, Nummer 5273, November 1970, S. 726–739, ISSN 0028-0836. PMID 5528785.
- L. Makowski, J. Bardhan u. a.: WAXS studies of the structural diversity of hemoglobin in solution. In: Journal of molecular biology. Band 408, Nummer 5, Mai 2011, S. 909–921, ISSN 1089-8638. doi:10.1016/j.jmb.2011.02.062. PMID 21420976. PMC 3081904 (freier Volltext).
- T. Yonetani, M. Laberge: Protein dynamics explain the allosteric behaviors of hemoglobin. In: Biochimica et biophysica acta. Band 1784, Nummer 9, September 2008, S. 1146–1158. doi:10.1016/j.bbapap.2008.04.025. PMID 18519045. PMC 2668241 (freier Volltext). (Review).
Weblinks
- Hämoglobin (Sauerstoffbindung, Aufbau) – Universität Saarland
- Interaktives 3D Modell von Hämoglobin – Proteopedia (engl.)
- Molekül des Monats: Hämoglobin (englisch)
- Jennifer McDowall/Interpro: Protein Of The Month: Haemoglobin. (engl.)